
Protenix-v1 Explained: An Open Biomolecular Structure Model Reaching AlphaFold-Class Accuracy with Transparent Benchmarks
Biomolecular structure prediction has rapidly evolved into one of the most important applications of deep learning in scientific research. For a long time, achieving state-of-the-art accuracy in predicting protein and complex molecular structures was associated with closed, highly controlled systems. That is beginning to change.
Table Of Content
A new project called Protenix-v1 demonstrates that it is possible to build an openly available system capable of predicting full atomic 3D structures of complex biomolecules at a level comparable to AlphaFold-class models — while releasing the entire pipeline, model weights, and evaluation tools to the public.
This is not just another model release. Protenix-v1 is an attempt to make high-accuracy biomolecular structure prediction transparent, reproducible, and extensible for researchers and developers.
What Exactly Is Protenix-v1?
Protenix-v1 is a foundation model designed to predict the 3D atomic structure of biomolecular complexes that may include:
Proteins
DNA
RNA
Small molecule ligands
Unlike many research models that only publish results, Protenix-v1 is released as a complete stack:
Training and inference code
Pretrained model weights
Data and multiple sequence alignment (MSA) pipelines
A browser-based web interface for interactive use
This means researchers are not limited to reading about the model’s performance — they can directly run it, inspect it, and modify it for their own experiments or production use.
Fair Comparison Through Matched Constraints
One of the most interesting aspects of Protenix-v1 is how its performance is evaluated. Instead of making vague claims about being “better” than existing models, the team ensured that comparisons were made under strictly aligned conditions.
The evaluation follows three important constraints that mirror AlphaFold3-style settings:
Same historical training data cutoff date
Comparable model scale
Similar inference and sampling budgets
By doing this, performance comparisons become meaningful. The model is not benefiting from newer data, larger compute budgets, or unfair evaluation advantages. It is tested in a way that allows researchers to understand how well it truly performs under equivalent conditions.
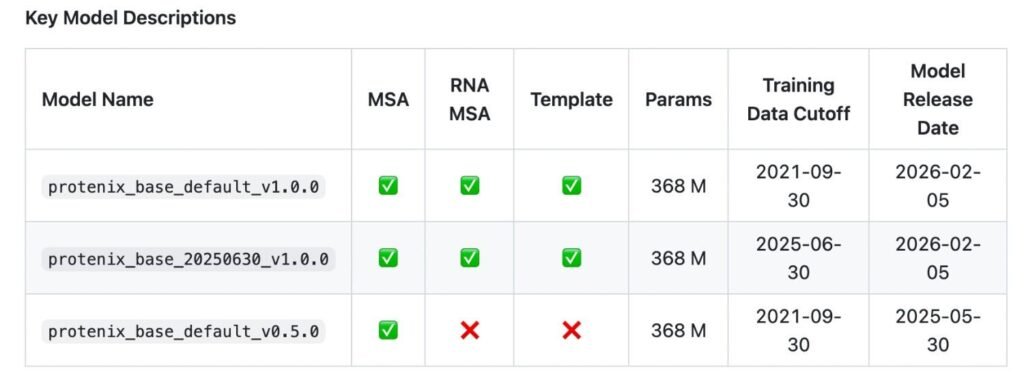
Introducing PXMeter: Benchmarking With Transparency
To support reproducible evaluation, the project also introduces PXMeter, a benchmarking toolkit created specifically for structure prediction tasks.
PXMeter provides:
A curated benchmark dataset covering thousands of biomolecular complexes
Carefully filtered entries to remove problematic or non-biological artifacts
Domain-specific and time-split evaluation subsets
Standardized metrics such as complex LDDT and DockQ
This is significant because benchmark design often affects how model performance is perceived. PXMeter attempts to standardize evaluation so that different models can be compared on the same ground without hidden biases.
Inference-Time Scaling: A Practical Accuracy Lever
Another notable feature of Protenix-v1 is how accuracy changes with increased sampling during inference.
Instead of having a single operating point, the model shows a predictable improvement in accuracy as more candidate structures are sampled. This creates a clear latency versus accuracy trade-off:
Fewer samples → faster predictions
More samples → higher structural accuracy
This gives users flexibility based on their computational budget and research needs.
Beyond Structure Prediction: A Growing Ecosystem
Protenix-v1 is part of a broader ecosystem of related tools that extend its usefulness:
Tools for protein–ligand docking using classical scoring approaches
Binder design systems built on top of the model
Lightweight compressed variants for faster inference with minimal accuracy loss
Because these tools share common formats and interfaces, they can be combined into larger research or production pipelines without significant integration effort.
Why This Release Matters
The importance of Protenix-v1 is not only about accuracy. It represents a shift toward:
Open reproducibility in scientific AI models
Transparent benchmarking practices
Modular systems that researchers can adapt and extend
Practical deployment options through web interfaces and lightweight variants
It lowers the barrier for universities, labs, and startups to experiment with high-quality biomolecular structure prediction without relying on closed systems.
Key Takeaways
Protenix-v1 is an open model for predicting full atomic 3D biomolecular structures.
It supports proteins, DNA, RNA, and small-molecule ligands.
Performance evaluation is done under carefully matched conditions for fairness.
PXMeter introduces transparent and standardized benchmarking.
The model demonstrates clear inference-time scaling behavior.
An ecosystem of docking, design, and lightweight variants extends its utility.
Final Thoughts
Protenix-v1 shows that high-level biomolecular structure prediction does not have to remain behind closed doors. By releasing the model, the pipelines, and the evaluation toolkit, the team has provided the research community with something far more valuable than a performance claim — a reproducible foundation.
For researchers working in structural biology, drug discovery, or computational chemistry, this opens up new opportunities to experiment, validate, and build upon an AlphaFold-class approach without restrictions.
As open science and AI continue to merge, projects like Protenix-v1 are likely to become increasingly important for the future of biomolecular research.